Aging affects reprogramming of murine pulmonary capillary endothelial cells after lung injury
Preprint, September 2024
https://www.biorxiv.org/content/10.1101/2023.07.11.548522v1Marin Truchi 1*, Grégoire Savary 2*, Marine Gautier-Isola 1, Hugo Cadis 1, Alberto Baeri 1, Arun Lingampally 3, Célia Scribe 1, Virginie Magnone 1, Cédric Girard-Riboulleau 1, Marie-Jeanne Arguel 1, Clémentine de Schutter 2, Julien Fassy 1, Nihad Boukrout 2, Romain Larrue 2, Nathalie Martin 2, Roger Rezzonico 1, Olivier Pluquet 2, Michael Perrais 2, Véronique Hofman 4, Charles-Hugo Marquette 5, Paul Hofman 4, Andreas Günther 6,7, Nicolas Ricard 8, Pascal Barbry 1, Sylvie Leroy 1,5, Kevin Lebrigand 1, Saverio Bellusci 3, Christelle Cauffiez 2, Georges Vassaux 1, Nicolas Pottier 2* and Bernard Mari 1*#
1Université Côte d'Azur, UMR CNRS 7275 Inserm 1323, IPMC, FHU-OncoAge, IHU RespiERA, Valbonne, France 2Univ. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, UMR9020 CNRS - U1277 Inserm - CANTHER, Lille, France. 3Excellence Cluster Cardio-Pulmonary System, German Center for Lung Research, Justus-Liebig-University Giessen, Giessen, Germany 4Université Côte d’Azur, Laboratory of Clinical and Experimental Pathology and Hospital-Integrated Biobank (BB-0033-00025), CHU Nice, FHU OncoAge, IHU RespiERA, Nice, France 5Département de Pneumologie, FHU-OncoAge, IHU RespiERA, CHU-Nice, France 6Center for Interstitial and Rare Diseases and Cardiopulmonary Institute, Justus-Liebig-University Gießen, Giessen, Germany 7European IPF Registry and Biobank 8Laboratory BioSanté U1292, Univ. Grenoble Alpes, INSERM, CEA, 38000, Grenoble, France
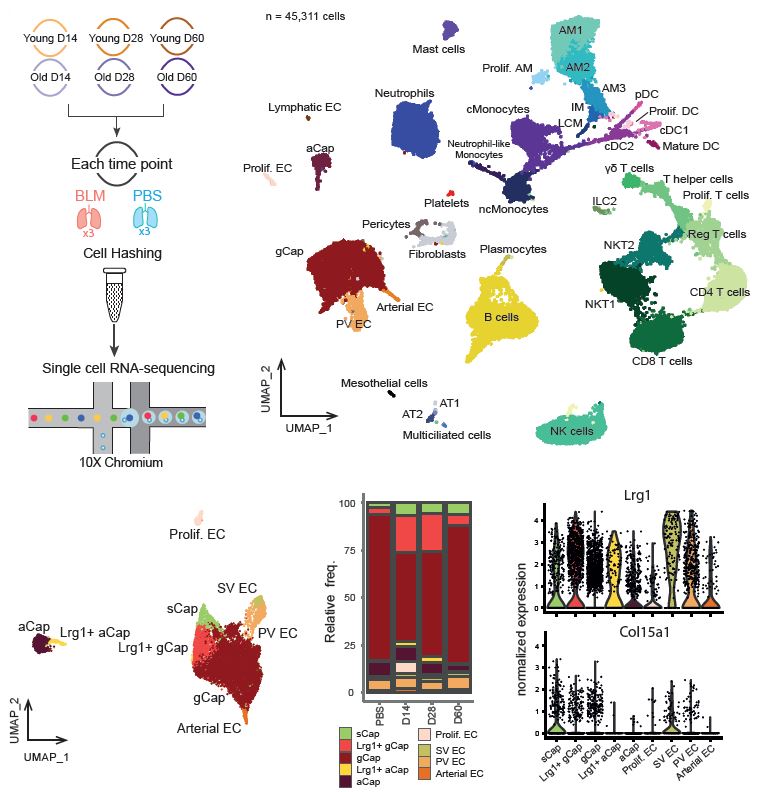
Aging increases the risk of developing fibrotic diseases by hampering tissue regeneration after injury. Using longitudinal single-cell RNA-seq and spatial transcriptomics, here we compare the transcriptome of bleomycin-induced fibrotic lungs of young and aged mice, at 3 time points corresponding to the peak of fibrosis, regeneration and resolution. We find that lung injury shifts the transcriptomic profiles of three pulmonary capillary endothelial cells (PCEC) subpopulations. The associated signatures are linked to pro-angiogenic signaling with strong Lrg1 expression and do not progress similarly throughout the resolution process between young and old animals. Moreover, part of this set of resolution-associated markers is also detected in PCEC from samples of patients with idiopathic pulmonary fibrosis (IPF). Finally, we find that aging also alters the transcriptome of PCEC which display typical pro-fibrotic and pro-inflammatory features. We propose that age-associated alterations in specific PCEC subpopulations may interfere with the process of lung progenitor differentiation, thus contributing to the persistent fibrotic process typical of human pathology.
Used pipelines
droplet-based single-cell RNA-seq
The analysis of single-cell gene expression data generated by droplet-based systems such as the 10X Genomics Chromium system involves several key steps. First, raw sequencing data are processed to generate gene expression matrices, which quantify the expression levels of genes in individual cells. Next, several data transformation steps are carried out to extract the biological variability that distinguishes communities of cells that share a common transcriptomic identity, reflecting their function or responses to stimuli. Finally, several statistical analyses can be carried out to quantify changes in tissue distribution or expression profiles for each of the populations identified, between different experimental conditions.
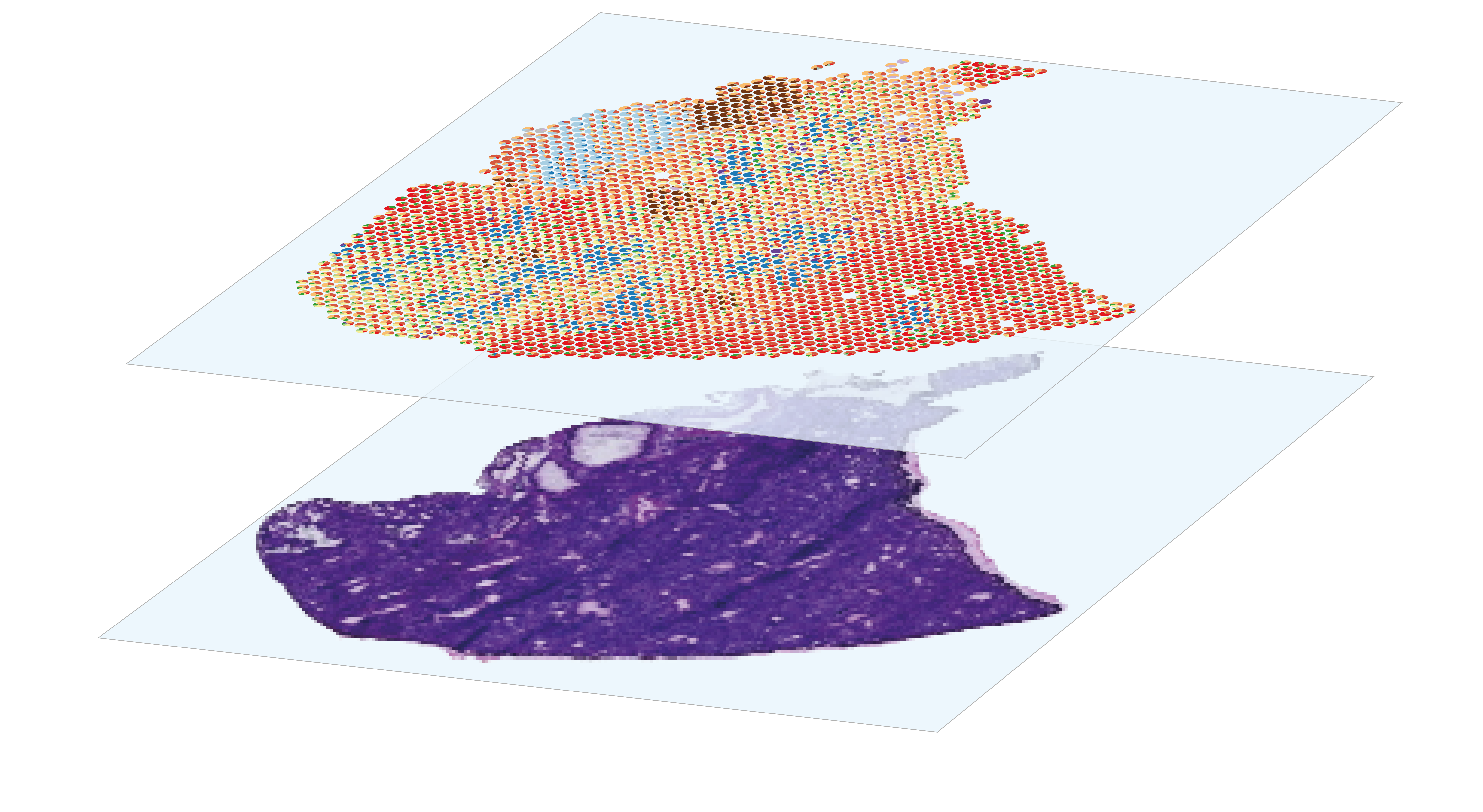
sequencing-based spatial transcriptomics
The analysis of spatial gene expression data generated by the Visium system begins with preprocessing the raw data with space ranger. Briefly, it consists in performing demultiplexing and quality control to filter out low-quality reads, aligning the remaining reads to a reference before counting gene expression from a single capture area. Once the matrix of gene counts per spot has been obtained, the aim is to identify the cell types represented in each spot. Although Visium technology avoids the dissociation step, its resolution is too low to perform single-cell characterization, as each 55µm diameter spot contains several cells. This implies that each spot is a combination of the transcriptome of potentially heterogeneous populations, which can be estimated using deconvolution methods, leveraging or not on a single-cell RNA-seq expression profils reference. After deconvolution, we can compare the distribution of cell types by histological region within the same section, or between sections from different experimental conditions. Combined with the spatial gene expression patterns, spots annotation can be used for Niche analysis, in order to characterize the interactions and relationships between cells and their microenvironment within tissues.
